
Harris Meyer: Amidst intense controversy, FDA approves Biogen’s Alzheimer's drug
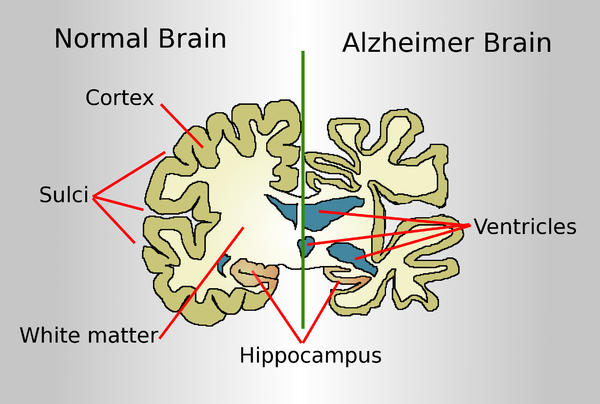
Drawing comparing a normal aged brain (left) and the brain of a person with Alzheimer's (right). Characteristics that separate the two are pointed out.
The Food and Drug Administration has approved the first treatment for Alzheimer’s disease, a drug developed by Biogen, which is based in Cambridge’s Kendall Square neighborhood. But the approval highlights a deep division over the drug’s benefits as well as criticism about the integrity of the FDA approval process.
The approval of aducanumab came despite a near-unanimous rejection of the product by an FDA advisory committee of outside experts in November. Doubts were raised when, in 2019, Biogen halted two large clinical trials of the drug after determining it wouldn’t reach its targets for efficacy. But the drugmaker later revised that assessment, stating that one trial showed that the drug reduced the decline in patients’ cognitive and functional ability by 22 percent.
Some FDA scientists in November joined with the company to present a document praising the intravenous drug. But other FDA officials and many outside experts say the evidence for the drug is shaky at best and that another large clinical trial is needed. A consumer advocacy group has called for a federal investigation into the FDA’s handling of the approval process for the product.
A lot is riding on the drug for Biogen. It is projected to carry a $50,000-a-year price tag and would be worth billions of dollars in revenue to the Cambridge company.
The FDA is under pressure because an estimated 6 million Americans are diagnosed with Alzheimer’s, a debilitating and ultimately fatal form of dementia, and there are no drugs on the market to treat the underlying disease. Although some drugs slightly mitigate symptoms, patients and their families are desperate for a medication that even modestly slows its progression.
Aducanumab helps the body produce antibodies that remove amyloid plaques from the brain, which has been associated with Alzheimer’s. It’s designed for patients with mild-to-moderate cognitive decline from Alzheimer’s, of which there are an estimated 2 million Americans. But it’s not clear whether eliminating the plaque improves brain function in Alzheimer’s patients. So far, nearly two dozen drugs based on the so-called amyloid hypothesis have failed in clinical trials.
Besides questions about whether the drug works, there also are safety issues. More than one-third of patients in one of the trials experienced brain swelling and nearly 20 percenty had brain bleeding, though those symptoms generally were mild and controllable. Because of those risks, patients receiving aducanumab have to undergo regular brain monitoring through expensive PET scans and MRI tests.
Some physicians who treat Alzheimer’s patients say they won’t prescribe the drug even if it’s approved.
“There’s a lot of hope among my patients that this is going to be a game changer,” said Dr. Matthew Schrag, an assistant professor of neurology at Vanderbilt University. “But the cognitive benefits of this drug are quite small, we don’t know the long-term safety risks, and there will be a lot of practical issues in deploying this therapy. We have to wait until we’re certain we’re doing the right thing for patients.”
Many aspects of aducanumab’s journey through the FDA approval process have been unusual. It’s “vanishingly rare” for a drug to continue on toward approval after its clinical trial was halted because unfavorable results showed that further testing was futile, said Dr. Peter Lurie, president of the Center for Science in the Public Interest and a former FDA associate commissioner. And it’s “mind-boggling,” he added, for the FDA to collaborate with a drugmaker in presenting a joint briefing document to an FDA advisory committee.
“A joint briefing document strikes me as completely inappropriate and an abdication of the FDA’s claim to being the best regulatory agency in the world,” Lurie said.
Three FDA advisory committee members who voted in November against approving the drug wrote in a recent JAMA commentary that the FDA’s “unusual degree of collaboration” with Biogen led to criticism that it “potentially compromised the FDA’s objectivity.” They cast doubt on both the drug’s safety and the revised efficacy data.
The FDA and Biogen declined to comment for this article.
Despite the uncertainties, the Alzheimer’s Association, the nation’s largest Alzheimer’s patient advocacy group, has pushed hard for FDA approval of aducanumab, mounting a major print and online ad campaign last month. The “More Time” campaign featured personal stories from patients and family members. In one ad, actor Samuel L. Jackson posted on Twitter, “If a drug could slow Alzheimer’s, giving me more time with my mom, I would have read to her more.”
But the association has drawn criticism for having its representatives testify before the FDA in support of the drug without disclosing that it received $525,000 in contributions last year from Biogen and its partner company, Eisai, and hundreds of thousands of dollars more in previous years. Other people who testified stated upfront whether or not they had financial conflicts.
Dr. Leslie Norins, founder of a group called Alzheimer’s Germ Quest that supports research, said the lack of disclosure hurts the Alzheimer’s Association’s credibility. “When the association asks the FDA to approve a drug, shouldn’t it have to reveal that it received millions of dollars from the drug company?” he asked.
But Joanne Pike, the Alzheimer’s Association’s chief strategy officer, who testified before the FDA advisory committee about aducanumab without disclosing the contributions, denied that the association was hiding anything or that it supported the drug’s approval because of the drugmakers’ money. Anyone can search the association’s website to find all corporate contributions, she said in an interview.
Pike said her association backs the drug’s approval because its potential to slow patients’ cognitive and functional decline offers substantial benefits to patients and their caregivers, its side effects are “manageable,” and it will spur the development of other, more effective Alzheimer’s treatments.
“History has shown that approvals of first drugs in a category benefit people because they invigorate the pipeline,” she said. “The first drug is a start, and the second and third and fourth treatment could do even better.”
Lurie disputed that. He said lowering the FDA’s standards and approving an ineffective or marginally effective drug merely encourages other manufacturers to develop similar, “me too” drugs that also don’t work well.
Anne Saint says she wouldn’t have risked putting her husband, Mike Saint, on the new Alzheimer’s drug aducanumab because of safety issues. Mike died in September at age 71. (MOLLY SAINT)
The Public Citizen Health Research Group, which opposes approval of aducanumab, has called for an investigation of the FDA’s “unprecedented and inappropriate close collaboration” with Biogen. It asked the inspector general of the Department of Health and Human Services to probe the approval process, which that office said it would consider.
The group also urged the acting FDA commissioner, Dr. Janet Woodcock, to remove Dr. Billy Dunn, an aducanumab advocate who testified about it to the advisory committee, from his position as director of the FDA’s Office of Neuroscience and hand over review of the drug to staffers who weren’t involved in the Biogen collaboration.
Woodcock refused, saying in a letter that FDA “interactions” with drugmakers make drug development “more efficient and more effective” and “do not interfere with the FDA’s independent perspective.”
Although it would be unusual for the FDA to approve a drug after rejection by an FDA advisory committee, it’s not unprecedented, Lurie said. Alternatively, the agency could approve it on a restricted basis, limiting it to a segment of the Alzheimer’s patient population and/or requiring Biogen to monitor patients.
“That will be tempting but shouldn’t be the way the problem is solved,” he said. “If the product doesn’t work, it doesn’t work. Once it’s on the market, it’s very difficult to get it off.”
If the drug is approved, Alzheimer’s patients and their families will have to make a difficult calculation, balancing the limited potential benefits with proven safety issues.
Anne Saint, whose husband, Mike, had Alzheimer’s for a decade and died in September at age 71, said that based on what she’s read about aducanumab, she wouldn’t have put him on the drug.
“Mike was having brain bleeds anyway, and I wouldn’t have risked him having any more side effects, with no sure positive outcome,” said Saint, who lives in Franklin, Tenn. “It sounds like maybe that drug’s not going to work, for a lot of money.”
Their adult daughter, Sarah Riley Saint, feels differently. “If this is the only hope, why not try it and see if it helps?” she said.
Harris Meyer is a Kaiser Health News reporter.

Llewellyn King: COVID-19 points way to faster medicines

In a Food and Drug Administration lab in Silver Spring, Md.
WEST WARWICK, R.I.
This is the month when the national spirit should start to lift: COVID-19 vaccines could be administered by mid-December. While we won’t reach the summit of a mighty mountain this month, nor well into next year, the ascent will have begun.
It is unlikely to be a smooth journey. There will be contention, accusation, litigation and frustration. Nothing so big as setting out to administer two-dose vaccines to the whole country could be otherwise.
But the pall that hangs so heavily over us with rising deaths, exhausted first responders and overstretched hospitals, will begin to lift very slightly.
For the rest of foreseeable history, there will be accusations leveled at the Trump administration for its handling of the pandemic — or its failure to handle it.
But one thing is certain: Our faith in our ability to make superhuman scientific efforts in the face of crisis will be restored. Developing a COVID-19 vaccine will be compared to putting a man on the Moon.
The large pharmaceutical companies, known collectively as Big Pharma, have shown their muscle. The lesson: Throw enough research and unlimited money at a problem, accelerate the regulatory process, and a solution can result.
Even globalization gets a good grade.
The first-to-market vaccine comes from American pharmaceutical giant Pfizer. But the vaccine was developed at its small German subsidiary, BioNTech, by a husband-and-wife team of first-generation Turkish immigrants. (Beware of whom you exclude.)
Biopharmaceutical research often takes place this way, akin to how it happens in Silicon Valley: Small companies innovate and invent, and larger ones gobble them up and provide the all-important resources for absurdly complicated and expensive clinical trials. These contribute mightily to the cost of new drugs. A new “compound” -- as a drug is called in the trade -- can cost up to $2 billion to bring to market; and financial reserves are needed, should there be costly lawsuits.
The development of new drugs looks like an inverted pyramid. Linda Marban, a researcher and CEO of Capricor Therapeutics Inc., a clinical-stage biotechnology company based in Los Angeles, explained it to me: “The last 20 years have shown a seismic change in how drugs and therapies are developed. Due to the speed at which science is advancing, and the difficulty of early-stage development, most of the early-stage work is done by small companies or the occasional academic. Big Pharma has moved into the role of late-stage clinical, sometimes Phase 2, but mostly Phase 3 and commercial development.”
In the upheaval occasioned by the pandemic, overhaul of the Food and Drug Administration looms large as a national priority. It must be able -- maybe with a greater use of artificial intelligence and data management -- to assess the safety and efficiency of desperately needed drugs without the current painful and often fatal delays.
Marban said of the FDA clinical-trials process: “It is the most laborious and frustrating process which delays important scientific and medical discoveries from patients. There are many situations where patients are desperate for therapy, but we have to climb the long and ridiculous ladder of doing clinical trials due to inefficiencies at the site which include nearly endless layers of contracting, budget negotiations, IRB [Institutional Review Board] approvals and, finally, interest and attention from overworked clinical trial staff.”
This situation, according to Marban, is compounded by the FDA’s requirement for clinical trials conducted and presented in a certain way, which often precludes getting an effective therapy to market. “If we simplify this process alone, we could move rapidly towards treatments and even cures for many horrific diseases,” she added.
War is a time of upheaval, and we are at war against the COVID-19. But war also involves innovation. We have proved that speed is possible when bureaucracy is energized and streamlined.
When COVID-19 is finally vanquished, it should leave a legacy of better medical research and sped-up approval procedures, benefiting all going forward.
Llewellyn King is executive producer and host of White House Chronicle, on PBS. His email is llewellynking1@gmail.com and he’s based in Rhode Island and Washington, D.C.
Linda Gasparello
Co-host and Producer
"White House Chronicle" on PBS
Mobile: (202) 441-2703
Websi
Liz Szabo/JoNel Alecia: Trump may try to rush in COVID-19 vaccine before election

It “seems frighteningly more plausible each day.’’
— Dr. Jerry Avorn, Harvard Medical School
President Trump, who seems intent on announcing a COVID-19 vaccine before Election Day, could legally authorize a vaccine over the objections of experts, officials at the Food and Drug Administration and even vaccine manufacturers, who have pledged not to release any vaccine unless it’s proved safe and effective.
In podcasts, public forums, social media and medical journals, a growing number of prominent health leaders say they fear that Trump — who has repeatedly signaled his desire for the swift approval of a vaccine and his displeasure with perceived delays at the FDA — will take matters into his own hands, running roughshod over the usual regulatory process.
It would reflect another attempt by a norm-breaking administration, poised to ram through a Supreme Court nominee opposed to existing abortion rights and the Affordable Care Act, to inject politics into sensitive public health decisions. Trump has repeatedly contradicted the advice of senior scientists on COVID-19 while pushing controversial treatments for the disease.
If the executive branch were to overrule the FDA’s scientific judgment, a vaccine of limited efficacy and, worse, unknown side effects could be rushed to market.
The worries intensified over the weekend, after Alex Azar, the administration’s secretary of Health and Human Services, asserted his agency’s rule-making authority over the FDA. HHS spokesperson Caitlin Oakley said Azar’s decision had no bearing on the vaccine approval process.
Vaccines are typically approved by the FDA. Alternatively, Azar — who reports directly to Trump — can issue an emergency use authorization, even before any vaccines have been shown to be safe and effective in late-stage clinical trials.
“Yes, this scenario is certainly possible legally and politically,” said Dr. Jerry Avorn, a professor of medicine at Harvard Medical School, who outlined such an event in the New England Journal of Medicine. He said it “seems frighteningly more plausible each day.”
Vaccine experts and public health officials are particularly vexed by the possibility because it could ruin the fragile public confidence in a COVID-19 vaccine. It might put scientific authorities in the position of urging people not to be vaccinated after years of coaxing hesitant parents to ignore baseless fears.
Physicians might refuse to administer a vaccine approved with inadequate data, said Dr. Preeti Malani, chief health officer and professor of medicine at the University of Michigan in Ann Arbor, in a recent Webinar. “You could have a safe, effective vaccine that no one wants to take.” A recent KFF poll found that 54 percent of Americans would not submit to a COVID-19 vaccine authorized before Election Day.
After this story was published, an HHS official said that Azar “will defer completely to the FDA” as the agency weighs whether to approve a vaccine produced through the government’s Operation Warp Speed effort.
“The idea the Secretary would approve or authorize a vaccine over the FDA’s objections is preposterous and betrays ignorance of the transparent process that we’re following for the development of the OWS vaccines,” HHS chief of staff Brian Harrison wrote in an email.
White House spokesperson Judd Deere dismissed the scientists’ concerns, saying Trump cared only about the public’s safety and health. “This false narrative that the media and Democrats have created that politics is influencing approvals is not only false but is a danger to the American public,” he said.
Usually, the FDA approves vaccines only after companies submit years of data proving that a vaccine is safe and effective. But a 2004 law allows the FDA to issue an emergency use authorization with much less evidence, as long as the vaccine “may be effective” and its “known and potential benefits” outweigh its “known and potential risks.”
Many scientists doubt a vaccine could meet those criteria before the election. But the terms might be legally vague enough to allow the administration to take such steps.
Moncef Slaoui, chief scientific adviser to Operation Warp Speed, the government program aiming to more quickly develop COVID-19 vaccines, said it’s “extremely unlikely” that vaccine trial results will be ready before the end of October.
Trump, however, has insisted repeatedly that a vaccine to fight the pandemic that has claimed 200,000 American lives will be distributed starting next month. He reiterated that claim Saturday at a campaign rally in Fayetteville, N.C.
The vaccine will be ready “in a matter of weeks,” he said. “We will end the pandemic from China.”
Although pharmaceutical companies have launched three clinical trials in the United States, no one can say with certainty when those trials will have enough data to determine whether the vaccines are safe and effective.
Officials at Moderna, whose vaccine is being tested in 30,000 volunteers, have said their studies could produce a result by the end of the year, although the final analysis could take place next spring.
Pfizer executives, who have expanded their clinical trial to 44,000 participants, boast that they could know their vaccine works by the end of October.
AstraZeneca’s U.S. vaccine trial, which was scheduled to enroll 30,000 volunteers, is on hold pending an investigation of a possible vaccine-related illness.
Scientists have warned for months that the Trump administration could try to win the election with an “October surprise,” authorizing a vaccine that hasn’t been fully tested. “I don’t think people are crazy to be thinking about all of this,” said William Schultz, a partner in a Washington, D.C., law firm who served as a former FDA commissioner for policy and as general counsel for HHS.
“You’ve got a president saying you’ll have an approval in October. Everybody’s wondering how that could happen.”
In an opinion piece published in The Wall Street Journal, conservative former FDA commissioners Scott Gottlieb and Mark McClellan argued that presidential intrusion was unlikely because the FDA’s “thorough and transparent process doesn’t lend itself to meddling. Any deviation would quickly be apparent.”
But the administration has demonstrated a willingness to bend the agency to its will. The FDA has been criticized for issuing emergency authorizations for two COVID-19 treatments that were boosted by the president but lacked strong evidence to support them: hydroxychloroquine and convalescent plasma.
Azar has sidelined the FDA in other ways, such as by blocking the agency from regulating lab-developed tests, including tests for the novel coronavirus.
Although FDA Commissioner Stephen Hahn told the Financial Times he would be willing to approve emergency use of a vaccine before large-scale studies conclude, agency officials also have pledged to ensure the safety of any COVID-19 vaccines.
A senior FDA official who oversees vaccine approvals, Dr. Peter Marks, has said he will quit if his agency rubber-stamps an unproven COVID-19 vaccine.
“I think there would be an outcry from the public health community second to none, which is my worst nightmare — my worst nightmare — because we will so confuse the public,” said Dr. Michael Osterholm, director of the Center for Infectious Disease Research and Policy at the University of Minnesota, in his weekly podcast.
Still, “even if a company did not want it to be done, even if the FDA did not want it to be done, he could still do that,” said Osterholm, in his podcast. “I hope that we’d never see that happen, but we have to entertain that’s a possibility.”
In the New England Journal editorial, Avorn and co-author Dr. Aaron Kesselheim wondered whether Trump might invoke the 1950 Defense Production Act to force reluctant drug companies to manufacture their vaccines.
But Trump would have to sue a company to enforce the Defense Production Act, and the company would have a strong case in refusing, said Lawrence Gostin, director of Georgetown’s O’Neill Institute for National and Global Health Law.
Also, he noted that Trump could not invoke the Defense Production Act unless a vaccine were “scientifically justified and approved by the FDA.”
Liz Szabo and JoNel Aleccia are Kaiser Health News reporters.
Liz Szabo: lszabo@kff.org, @LizSzabo
JoNel Aleccia: jaleccia@kff.org, @JoNel_Aleccia
N.E. Council update on COVID-19-related activities

“Brother Jonathan’’ was a 19th Century personification of New England.
BOSTON
COVID-19 briefing from The New England Council (newenglandcouncil.com)
“As our region and our nation continue to grapple with the Coronavirus Disease (COVID-19) pandemic, The New England Council is using our blog as a platform to highlight some of the incredible work our members have undertaken to respond to the outbreak. Each day, we’ll post a round-up of updates on some of the initiatives underway among Council members throughout the region. We are also sharing these updates via our social media, and encourage our members to share with us any information on their efforts so that we can be sure to include them in these daily round-ups.
You can also check our COVID-19 Virtual Events Calendar for information on upcoming COVID-19 related programming – including Congressional town halls and webinars presented by NEC members.
Here is the March 30 roundup:
Medical Response
Novartis Anti-Malaria Drug Gets Emergency Approval from FDA – Following emergency approval from the Food and Drug Administration (FDA) of two anti-malaria drugs for the potential treatment of COVID-19, Novartis, along with Bayer, is contributing millions of doses of the drugs, hydroxychloroquine and chloroquine. The drugs have previously received much attention following some anecdotal evidence to support the benefits to relieve respiratory symptoms of infected patients, and could be a potential treatment to reduce symptoms and strain on healthcare providers. The Washington Post has more.
Sanofi Expands Clinical Trial – After announcing the beginnings of its clinical trials in the United States last week, Sanofi—along with Regeneron Pharmaceuticals—has expanded the trial in Italy, Spain, Germany, France, Canada, and Russia. The trials will test the rheumatoid arthritis drug Kevzara as a potential treatment for patients around the world. Read more in CNBC.
Economic/Business Continuity Response
New Balance Shifts to Producing Masks – At its Lawrence, Mass., manufacturing facility, New Balance has begun producing prototypes for face masks to distribute around the region and country. As hospitals and those exposed to the virus begin to see shortages of protective gear such as face masks, New Balance has joined other local manufacturers in an “all hands on deck” response to the need. Read more in WCVB Boston.
Harvard University Guarantees Pay for All Workers Through End of Semester – To ensure that those who are willing and able to work are paid, Harvard University updated its policies to include payment and benefits for all workers through May 28. The updated policy includes those employed in dining, security, and custodial services across the university, as well as those in administrative and contract roles. Harvard is also providing stabilization funds to the six independent childcare centers on campus to allow the employees to work. The Boston Business Journal
Community Response
Suffolk University, Pine Street Inn to Repurpose University Residence Hall to House Homeless Population – In light of its switch to remote learning, Suffolk University has offered a residence hall to the homeless population of Boston. The residence hall will provide 172 beds, and will be managed by both Pine Street Inn and the city’s Public Health Commission to reduce congestion and increase social distancing in shelters. The Boston Business Journal has more.
AARP Launches Platform to Connect Volunteers with Those in Need – AARP announced its new online platform, AARP Community Connections, to organize and mobilize volunteer groups and connect them with people and families who need assistance. From groceries to financial assistance to emotional support, users of the platform can receive a wide variety of support in the age of social distancing. Read more in KTVZ.
Amgen Donates $12.5 Million to Relief Efforts – Amgen has committed $12.5 million to support the efforts of local emergency and patient-focused services. In addition to the funds, Amgen is supporting online learning platforms to provide resources to students as they transition to remote instruction. Read more.
Tufts Health Plan Announces $500,000 for Food-Insecure Elderly People – Tufts Health Plan has committed the first half of its $1 million donation in relief efforts to combat food insecurity among in the New England region. Tufts has identified 21 organizations that provide meals and other support to individuals we well as food banks and other businesses coordinating a regional response as the recipients of the first wave of funding. Read more in the Portsmouth Press.
Stay tuned for more updates each day, and follow us on Twitter for more frequent updates on how Council members are contributing to the response to this global health crisis.’’

Chris Powell: Plenty of voter fraud in Bridgeport; piling on Purdue Pharma

Iranistan, the residence of P.T. Barnum, in 1848
According to Connecticut Secretary of the State Denise Merrill, the Nutmeg State has too little voter fraud to worry about. But she doesn't really know, because until last week few people had ever seriously looked.
But last week Connecticut's Hearst newspapers looked into the extraordinary level of absentee voting in Bridgeport's recent Democratic mayoral primary election, in which the challenger, state Sen. Marilyn Moore, won on the voting machines but was defeated as Mayor Joe Ganim overwhelmingly carried the absentee ballots.
The Hearst investigation found that fraud was extensive among its limited sample of voters. Ineligible people -- including people who were not registered to vote, people who were not Democrats, and felons and parolees -- received and cast absentee votes. Elderly people were coerced or pressured to complete absentee ballots for the mayor by Ganim supporters who came to their homes. Absentee ballots were sent to people who did not request them. Record-keeping by Bridgeport election officials is sloppy, maintaining incorrect birthdates for some voters and mistaken receipt dates for absentee ballots.
Secretary Merrill has forwarded the Hearst report to the state Elections Enforcement Commission and asked it to investigate because her office lacks the commission's powers. But the secretary should be chastened by what already has come out, for she has been advocating legislation to deny public access to voter registration data
With her legislation Merrill claims to be supporting individual privacy. But voters are not entirely private citizens, for they hold the most basic public office -- elector -- an office established by the state Constitution. Nobody has to become an elector. You volunteer, and election fraud cannot be detected by the public or news organizations unless the names, addresses, and birthdates of electors are as public as they long have been in Connecticut.
Since, as her legislation signifies, the secretary denies the possibility of voter fraud, the law should not hinder the press and public in detecting it as the Hearst papers have just done.
* * *
PILING ON PURDUE PHARMA: If there was an award for piling on, Connecticut Atty. Gen. William Tong would be a leading contender. Practically every day he announces a lawsuit his office is joining to challenge some policy of the Trump administration.
Those policies may be questionable but it is also questionable how much Tong's office is really doing with the lawsuits beyond providing pro-forma endorsements that get publicity for him.
Tong has worked up his greatest indignation for the lawsuit he has joined with many states against Stamford-based Purdue Pharma, manufacturer of the painkiller OxyContin, to which many people have gotten addicted, many of them dying from their addiction. Tong wants the company liquidated and the proceeds somehow distributed to the drug's supposed victims.
But the country's worsening addiction problem long preceded OxyContin, and nobody could have gotten addicted to it if the U.S. Food and Drug Administration hadn't approved it 24 years ago and if thousands of doctors had not prescribed it too heavily to their patients. The FDA and those doctors bear the immediate responsibility for abuse of the drug, not the manufacturer, since from the beginning OxyContin has been a controlled substance.
Of course suing those who uncontrolled the drug would be a tougher and fairer fight for any attorney general who enjoys piling on.
Chris Powell is a columnist for the Journal Inquirer, in Manchester, Conn.
Anna Meyer: Your tax dollars fund agribusiness propaganda

Via OtherWords.org:
While Congress hasn’t accomplished much in 2017, it did manage to pass a budget resolution — and within that budget, a sum of $3 million stands out.
Congress appropriated that $3 million to fund the Agricultural Biotechnology Education and Outreach Initiative. That’s a partnership between the Food & Drug Administration (FDA) and the Department of Agriculture (USDA) “to provide consumer education on agricultural biotechnology and food and animal feed ingredients derived from biotechnology.”
What they’re really talking about is a promotional campaign for genetically modified organisms, or GMOs.
There are two major flaws with this plan.
First, the FDA is tasked with building a campaign around the “safety and benefits of crop biotechnology.” But what about the risks, concerns, and unknowns?
Leaving those out means using government agencies and taxpayer funds for corporate propaganda. It benefits companies like Monsanto, Dow, Dupont, Syngenta and Bayer, which collectively earn billions of dollars from these technologies, but does little to inform consumers.
Second, the initiative will push forward “science-based” education. The question is: Whose science are they using?
There’s very little independent or government research on GMOs and their corresponding pesticides. The lack of unbiased and comprehensive science on biotechnology is a result of corporations controlling who can do research on biotech products.
Much of the existing research is either industry-funded or straight out of biotechnology companies’ own labs. The existing regulatory framework relies on voluntary reporting and doesn’t require independent verification to prove the safety of new products before they land on dinner plates across the country.
If the government’s going to educate consumers on biotechnology, it must first do its own unbiased studies on the long-term environmental and health impacts of existing GMOs and pesticides. It also needs a much more rigorous — and mandatory — regulatory process.
The government must tell consumers the full truth, presenting balanced and unbiased information on the benefits, risks, and concerns around biotechnology. The FDA must openly address consumer concerns about long-term environmental impacts, corporate influence on government research, and corporate control of our industrialized food system.
We’re at a turning point in history where we can reverse the harm that we’ve done to our communities, farmland, and environment.
Industrialized, chemical-intensive agriculture designed to work around biotechnology is a failed system. It’s increasing herbicide use, exacerbating pesticide resistance, polluting our waterways, soil, and air, and promoting highly processed food and confined animal production.
In order to build a more sustainable food system for our health and our climate, we need to move away from chemical-intensive agriculture. Rather than promoting corporate interests, that $3 million would be much better used to promote the transition to regenerative organic agriculture, to build urban food hubs, and to aid the next generation of farmers in accessing land and resources.
The FDA doesn’t need a biotechnology marketing initiative. It needs an initiative to bring back public trust in federal regulatory agencies, and move the country forward towards truly sustainable agriculture.
Anna Meyer is the Food Campaigns Fellow at Green America.